
Cardiac amyloidosis is a progressive, infiltrative cardiomyopathy in which there are abnormal amyloid fibril deposits in the interstitial space between cardiac myocytes. Over time, this infiltration leads to progressive diastolic dysfunction with a restrictive pattern and prominent right ventricular failure. Managing cardiac amyloidosis is challenging and complex because patients often experience refractory symptoms, medication intolerance, multisystem involvement, and hypotension. Although amyloidosis was thought to be rare, improved diagnostic studies, better recognition of symptoms, and the availability of newer therapies have led to increased diagnosis and incidence.
As new targeted treatments become available, including those that improve neurologic symptoms, early identification of amyloidosis is important to delay disease progression and improve quality of life. Several national and international societies, including the American College of Cardiology and American Heart Association, have issued scientific statements to guide diagnosis and management of this disease.
Here are five things to know about the diagnostic workup of cardiac amyloidosis.
1. Disease diagnosis requires a high index of suspicion in the presence of specific clinical manifestations.
Although cardiac amyloidosis cannot be diagnosed through routine testing, there are several clinical features or "red flags" that should raise suspicion for amyloidosis and which warrant further testing. Clinical manifestations of cardiac amyloidosis include cardiac (eg, heart failure, atrial fibrillation, bradyarrhythmias, conduction abnormalities, pacemakers), musculoskeletal (carpal tunnel syndrome, back pain or lumbar stenosis, ruptured distal biceps tendon), polyneuropathic (neuropathy in the hands and feet, muscle weakness, difficulty ambulating, falls), and autonomic dysfunction (orthostatic hypotension, intolerance to blood pressure medications, chronic diarrhea, chronic constipation, unintended weight loss, erectile dysfunction) symptoms.
In addition to clinical manifestations, persistently elevated serum troponin and B-type natriuretic peptide (BNP) levels are frequently seen in cardiac amyloidosis; however, these biomarkers are not specific for amyloid. Troponin and BNP levels within the normal range are quite sensitive in excluding the diagnosis of amyloidosis.
The presence of cardiac and extracardiac manifestations warrant specific laboratory testing and imaging studies. Electrocardiography (ECG) and transthoracic echocardiography are initial imaging studies in the diagnostic workup of patients with suspected cardiac amyloidosis. Common findings on ECG that increase suspicion for cardiac amyloidosis include low QRS voltage, atrial fibrillation, conduction system disease, and pseudoinfarct pattern. Multiple features on echocardiography suggestive of cardiac amyloidosis include increases in left ventricular (LV) wall thickness (> 1.2 cm), echogenicity of the myocardium, thickening of the interatrial septum and valves (> 0.5 cm), and relative LV wall thickness (> 0.42 cm); grade 2 or higher diastolic dysfunction; reduced tissue Doppler velocities; "cherry-on-the-top" sign on speckle-tracking longitudinal strain bullseye map; atrial enlargement and dysfunction; pericardial effusion; increases in pulmonary artery and right atrium pressures; and decreased global longitudinal LV strain (absolute value < 15%). Multiple features of amyloidosis seen on these diagnostic studies warrant further specialized testing.
2. There are two main types of cardiac amyloidosis.
Although there are more than 36 types of amyloid precursor proteins, only nine build up in the myocardium and cause cardiac amyloidosis. The different types of amyloidosis are named by the letter "A" followed by an abbreviation for the type of precursor protein that has misfolded. The most common forms of cardiac amyloidosis are immunoglobulin light chain amyloidosis (AL-CM) and transthyretin amyloidosis (ATTR-CM).
In AL amyloidosis, plasma cell dyscrasias produce kappa or lambda monoclonal light chains as an intact molecule or fragment, though the lambda isotope occurs in approximately 75%-80% of cases. The light chain misfolds and forms a beta-pleated sheet instead of an alpha-helical chain. After an increase in serum levels of the free chains, the protein deposits in organs, causing significant dysfunction. The estimated annual incidence and prevalence of AL amyloidosis is approximately 1 in 75,000-100,000 and 1 in 25,000, respectively.
ATTR amyloidosis develops when misfolded transthyretin proteins, which are primarily synthesized by the liver, accumulate in the myocardium. There are two subtypes of ATTR amyloidosis: wild-type (wtATTR) and hereditary ATTR, also referred to as variant ATTR (ATTRv). The misfolding or aggregating TTR protein is genetically normal in patients with wtATTR, whereas there is a mutation that makes the TTR protein more prone to misfolding in ATTRv. The true prevalence of wtATTR amyloidosis is not known, though it is significantly more common than previously thought. On the basis of data from Cuscaden and colleagues, global cases of wtATTR were estimated to be about 8.5 million in 2019 and are expected to rise to nearly 25 million by 2050.
3. When cardiac amyloidosis is suspected, the first step is to assess for monoclonal protein.
It is important to determine the presence of a monoclonal gammopathy in the amyloidosis workup. Concomitant measurement of urine and serum immunofixation electrophoresis along with serum free light chains has a sensitivity > 99%; therefore, these laboratory tests should be done together. The ratio of kappa and lambda free light chains is particularly important in patients with chronic kidney disease as clearance of the free chains occurs via the reticuloendothelial system and kidneys. Given that kappa free light chains are made at a higher rate than lambda free light chains, the kidneys are generally more efficient at clearing kappa free light chains. Increased kappa or lambda levels are common in patients with kidney disease. In the setting of an abnormal monoclonal protein result, collaboration with a hematologist is recommended to determine the underlying cause.
After abnormal monoclonal antibody identification, a tissue diagnosis is required for AL amyloidosis. This can be done with bone marrow or fat pad biopsy; however, performing bone marrow and fat pad biopsies at the same time increases sensitivity (80%-90%) and usually avoids the need for organ biopsy. Although monoclonal gammopathy is suggestive of AL amyloidosis, monoclonal gammopathy of unknown significance (MGUS) can also occur with ATTR amyloid and other less common types of amyloidosis.
4. Cardiac scintigraphy should be performed when evaluating for ATTR-CM.
Multiple tracers can be used for cardiac scintigraphy, including 99mtechnetium pyrophosphate (99mTc-PYP), 99mTc 3,3-diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD), and 99mTc-hydroxymethylene diphosphonate (99mTc-HMDP). In the United States, the only agent readily available for identifying ATTR amyloidosis is 99mTC-PYP. Imaging should be performed with planar and single-photon emission CT to demonstrate diffuse myocardial uptake and differentiate this from blood pool uptake.
A qualitative and semi-quantitative scoring system has been developed on the basis of uptake of the radiotracer in order to make the diagnosis of ATTR-CM. The qualitative score compares the relative tracer uptake in the myocardium relative to the ribs and grades it using this scale:
Grade 0: no myocardial uptake and normal bone uptake
Grade 1: mild myocardial uptake, less than rib uptake
Grade 2: moderate myocardial uptake, equal to rib uptake
Grade 3: severe myocardial uptake, greater than rib uptake
Quantitative scores are based on the heart-to-contralateral lung uptake ratio with 99mTc-PYP tracers or the heart-to-whole body ratio with 99mTc-DPD and 99mTc-HMDP tracers. Because the isotope in the myocardium dissipates over time, a positive test for ATTR-CM is 1.3 at 3 hours and 1.5 at 1 hour. A diagnosis of cardiac amyloidosis cannot be made solely on the basis of the heart-to-contralateral chest ratio. In the absence of a clonal disorder, grade 2 or 3 myocardial uptake of 99mTC-PYP is diagnostic for ATTR-CM.
5. Diagnostic tools for cardiac amyloidosis include cardiac magnetic resonance imaging, cardiac biopsy, and genetic testing.
Cardiac magnetic resonance (CMR) imaging can be considered in the work up for cardiac amyloidosis in setting of suggestive or indeterminate findings on monoclonal protein evaluation or on 99mTc-PYP scan. Typical CMR findings of patients with biopsy-confirmed systemic amyloidosis include increased myocardial wall thickness, increased myocardial mass, diffuse late gadolinium enhancement, nulling of the myocardium before or at the same inversion time as the blood pool, and extensive extracellular volume expansion. If none of these findings are present, cardiac amyloidosis is less likely. With positive findings, a cardiac biopsy can be considered to confirm the diagnosis.
Endomyocardial biopsy, the gold standard for diagnosing cardiac amyloidosis, is typically recommended in the setting of a positive 99mTc-PYP scan and concomitant abnormal monoclonal gammopathy test result, an equivocal 99mTc-PYP scan in the setting of high clinical suspicion, and when 99mTc-PYP scan is not available. In all patients with suspected AL amyloidosis, a tissue biopsy is necessary to determine the presence and type of amyloid. Biopsy of the clinically affected organ, however, is not always necessary. Biopsy of the iliac crest bone marrow along with abdominal subcutaneous fat aspiration or punch biopsy can identify amyloid in 85% of cases. If this fails to identify amyloid protein and clinical suspicion remains high, an endomyocardial biopsy can be undertaken. Bone marrow biopsy and fat pad biopsy, however, have poor sensitivity for ATTR amyloidosis.
Typically, on the biopsy samples, amyloid fibrils are identified by positive Congo red uptake with green birefringence under polarized light. Tissue is then further characterized by immunohistochemistry or mass spectrometry–based analysis. The sensitivity and specificity for amyloid typing with immunohistochemistry is low; however, a study found that combined specific sampling by laser microdissection and tandem mass spectrometry had a sensitivity and specificity of 98% in identifying amyloid type in the validation set. This is necessary to identify the amyloidogenic precursor to further delineate appropriate treatment.
Genetic testing is indicated when ATTR amyloidosis is identified. TTR is encoded by a single-copy gene TTR on chromosome 18 and is inherited in an autosomal-dominant pattern. It is appropriate to test the proband (first member of a family to be identified) when a cardiac amyloidosis phenotype has been established; this is necessary in finding a more relevant variant that is pathogenic or disease-associated.
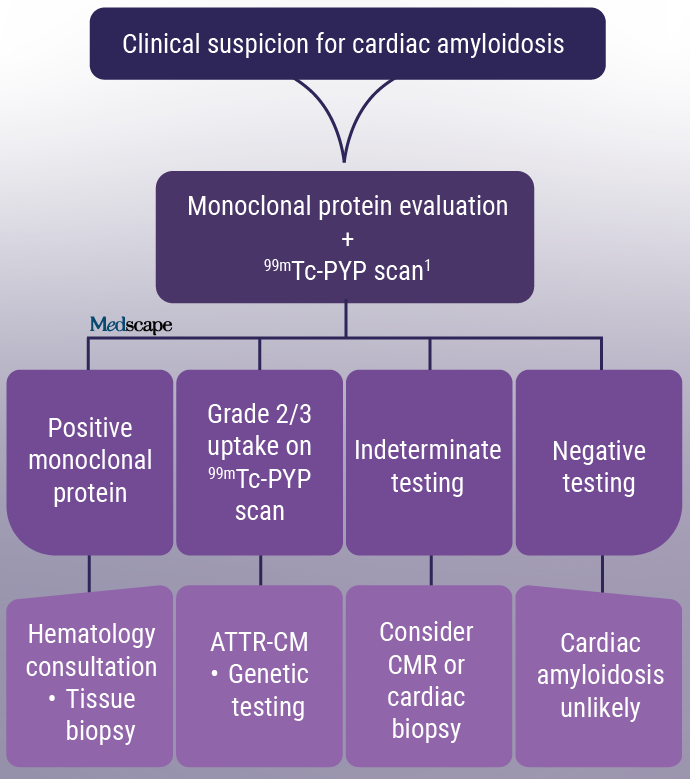
Figure. Diagnostic pathway for cardiac amyloidosis (modified). CMR= cardiac magnetic resonance; 99mTC-PYP = Technetium pyrophosphate scintigraphy scan. 1 99mTc-PYP scan may be ordered simultaneously, but must be interpreted in the context of a negative monoclonal protein screen.
In summary, early detection of cardiac amyloidosis and prompt initiation of appropriate therapy are critical to improve long-term outcomes. Because of the complex nature and involvement of multiple organ systems, diagnosis of this disease requires a high clinical suspicion. In these instances, following the current diagnostic approach (Figure), including laboratory evaluation and imaging studies, can help better patients' quality of life.
Follow Medscape on Facebook, X (formerly known as Twitter), Instagram, and YouTube
Credits:
Lead image: iStock/Getty Images
Cite this: Diagnosing Cardiac Amyloidosis: 5 Things to Know - Medscape - Nov 20, 2023.








Comments